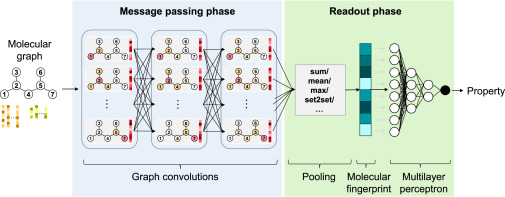
Abstract
Graph neural networks (GNNs) are emerging in chemical engineering for the end-to-end learning of physicochemical properties based on molecular graphs. A key element of GNNs is the pooling function which combines atom feature vectors into molecular fingerprints. Most previous works use a standard pooling function to predict a variety of properties. However, unsuitable pooling functions can lead to unphysical GNNs that poorly generalize. We compare and select meaningful GNN pooling methods based on physical knowledge about the learned properties. The impact of physical pooling functions is demonstrated with molecular properties calculated from quantum mechanical computations. We also compare our results to the recent set2set pooling approach. We recommend using sum pooling for the prediction of properties that depend on molecular size and compare pooling functions for properties that are molecular size-independent. Overall, we show that the use of physical pooling functions significantly enhances generalization.