Insight to gene expression from promoter libraries with the machine learning workflow Exp2Ipynb
Abstract
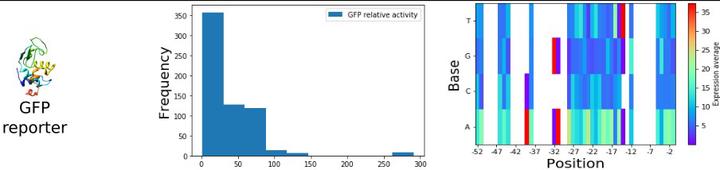
Metabolic engineering relies on modifying gene expression to regulate protein concentrations and reaction activities. The gene expression is controlled by the promoter sequence, and sequence libraries are used to scan expression activities and to identify correlations between sequence and activity. We introduce a computational workflow called Exp2Ipynb to analyze promoter libraries maximizing information retrieval and promoter design with desired activity. We applied Exp2Ipynb to seven prokaryotic expression libraries to identify optimal experimental design principles. The workflow is open source, available as Jupyter Notebooks and covers the steps to 1) generate a statistical overview to sequence and activity, 2) train machine-learning algorithms, such as random forest, gradient boosting trees and support vector machines, for prediction and extraction of feature importance, 3) evaluate the performance of the estimator, and 4) to design new sequences with a desired activity using numerical optimization. The workflow can perform regression or classification on multiple promoter libraries, across species or reporter proteins. The most accurate predictions in the sample libraries were achieved when the promoters in the library were recognized by a single sigma factor and a unique reporter system. The prediction confidence mostly depends on sample size and sequence diversity, and we present a relationship to estimate their respective effects. The workflow can be adapted to process sequence libraries from other expression-related problems and increase insight to the growing application of high-throughput experiments, providing support for efficient strain engineering.