Machine learning and molecular descriptors enable rational solvent selection in asymmetric catalysis
Abstract
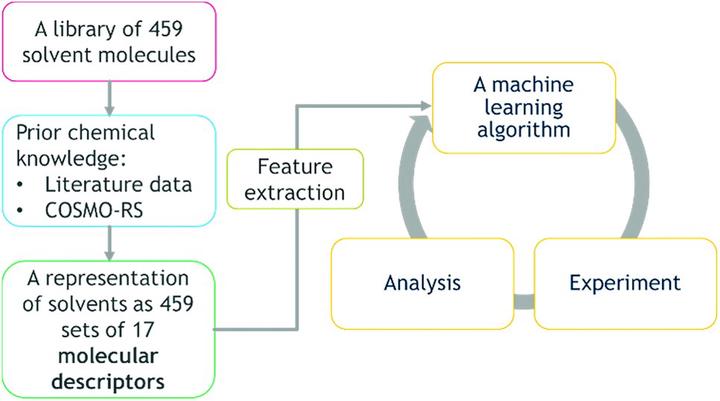
Rational solvent selection remains a significant challenge in process development. Here we describe a hybrid mechanistic-machine learning approach, geared towards automated process development workflow. A library of 459 solvents was used, for which 12 conventional molecular descriptors, two reaction-specific descriptors, and additional descriptors based on screening charge density, were calculated. Gaussian process surrogate models were trained on experimental data from a Rh(CO)2(acac)/Josiphos catalysed asymmetric hydrogenation of a chiral α-β unsaturated γ-lactam. With two simultaneous objectives – high conversion and high diastereomeric excess – the multi-objective algorithm, trained on the initial dataset of 25 solvents, has identified solvents leading to better reaction outcomes. In addition to being a powerful design of experiments (DoE) methodology, the resulting Gaussian process surrogate model for conversion is, in statistical terms, predictive, with a cross-validation correlation coefficient of 0.84. After identifying promising solvents, the composition of solvent mixtures and optimal reaction temperature were found using a black-box Bayesian optimisation. We then demonstrated the application of a new genetic programming approach to select an appropriate machine learning model for a specific physical system, which should allow the transition of the overall process development workflow into the future robotic laboratories.